¿Por qué el gen letal de Huntington tarda tanto en causar daño?
Síntomas típicos de la enfermedad: movimientos involuntarios, deterioro cognitivo y cambios de comportamiento

Fact checked
Este artículo de OkSalud ha sido verificado para garantizar la mayor precisión y veracidad posible: se incluyen, en su mayoría, estudios médicos, enlaces a medios acreditados en la temática y se menciona a instituciones académicas de investigación. Todo el contenido de OkSalud está revisado pero, si consideras que es dudoso, inexacto u obsoleto, puedes contactarnos para poder realizar las posibles modificaciones pertinentes.
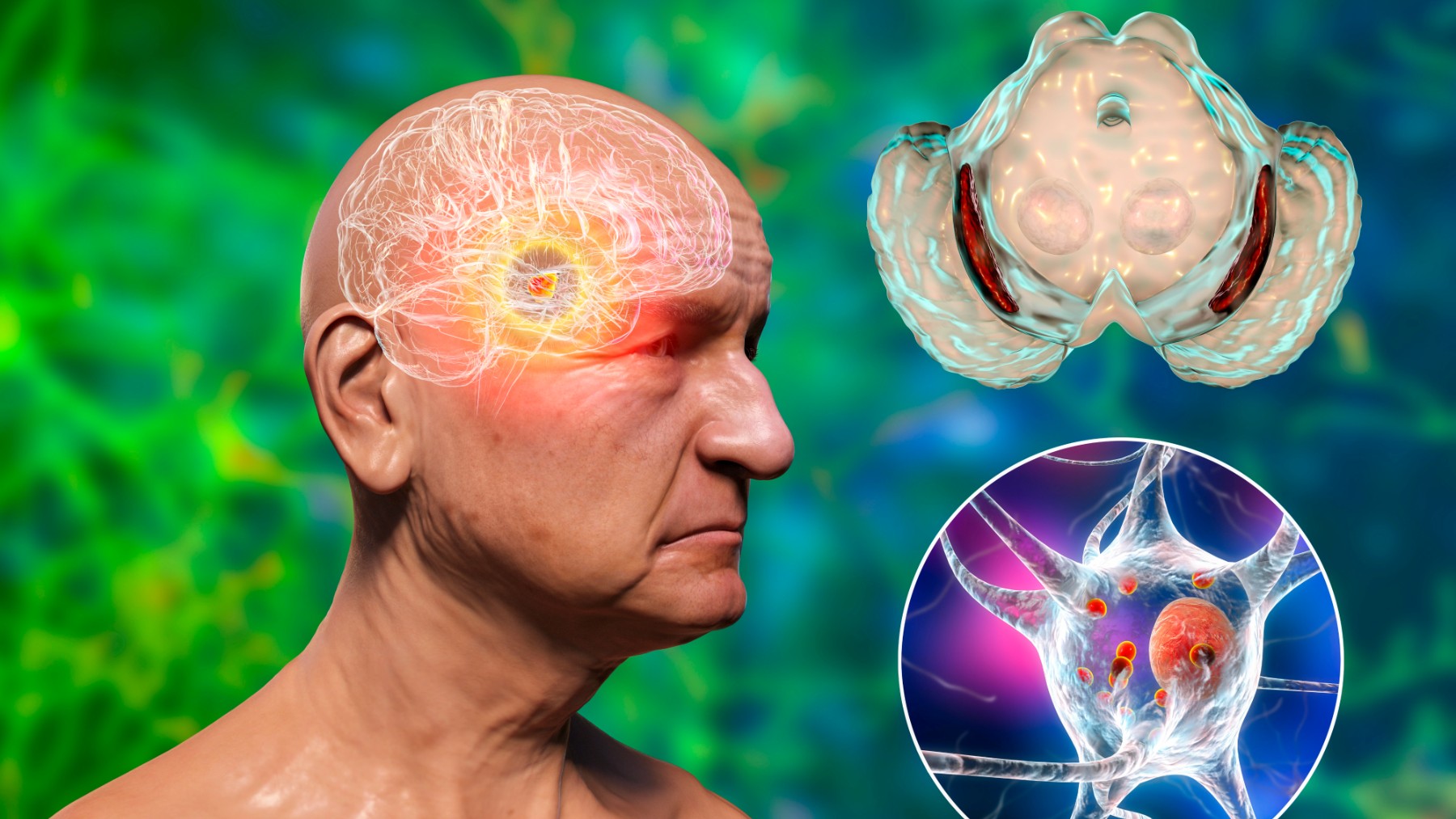
La enfermedad de Huntington es una condición neurológica devastadora, conocida por ser hereditaria y mortal. Este trastorno, causado por un gen defectuoso en el cromosoma 4, afecta a millones de personas en todo el mundo, pero lo más intrigante es su lento progreso: los síntomas suelen aparecer entre los 30 y 50 años, mucho después de que el daño genético ya esté presente. ¿Qué es lo que retrasa su manifestación?
La enfermedad de Huntington es causada por una mutación en el gen HTT, que codifica la proteína huntingtina. Esta mutación provoca una expansión anormal de repeticiones del trinucleótido CAG, que genera una versión alterada y tóxica de la proteína. Con el tiempo, esta huntingtina defectuosa se acumula en las neuronas, formando agregados que interfieren con su función normal.
Sin embargo, los estudios han revelado que esta acumulación no ocurre de la noche a la mañana. En las primeras etapas de la vida, los mecanismos celulares de limpieza, como la autofagia y los proteasomas, logran controlar los niveles de proteína defectuosa. Pero con el paso de las décadas, estas defensas comienzan a fallar, lo que permite que los agregados se acumulen y dañen las células nerviosas. Y es que, como se publica en Nature, una mutación en las neuronas crece durante décadas antes de alcanzar un límite mortal
Factores protectores
Un factor clave en el retraso de los síntomas es la resiliencia inicial del cerebro. Durante las primeras etapas de la vida, las neuronas cuentan con una capacidad impresionante para compensar el daño. Los científicos han identificado varios factores que contribuyen a esta resistencia:
- Plasticidad neuronal: el cerebro joven puede reorganizarse y adaptarse para minimizar el impacto de las neuronas disfuncionales.
- Sistemas de reparación: las células tienen mecanismos avanzados de reparación de ADN y control de calidad de proteínas, que son más eficaces en la juventud.
- Proteínas chaperonas: estas moléculas ayudan a plegar correctamente las proteínas y a prevenir la formación de agregados tóxicos.
Con el envejecimiento, estos sistemas comienzan a deteriorarse, dejando al cerebro vulnerable a los efectos acumulativos de la huntingtina mutada.
Envejecimiento
El envejecimiento actúa como un catalizador en la enfermedad de Huntington. Estudios recientes han mostrado que los cambios en las mitocondrias, los orgánulos responsables de producir energía en las células, desempeñan un papel fundamental. Con el tiempo, la huntingtina mutada daña las mitocondrias, lo que lleva a una disminución en la energía disponible para las neuronas y a un aumento del estrés oxidativo.
Además, la inflamación crónica en el cerebro, provocada por la respuesta inmunitaria activada por la proteína mutada, acelera la muerte celular. Este proceso es gradual, pero se intensifica con los años, culminando en los síntomas típicos de la enfermedad: movimientos involuntarios, deterioro cognitivo y cambios de comportamiento.
Implicaciones para el tratamiento
Comprender por qué el gen letal de Huntington tarda tanto en causar daño abre nuevas posibilidades para el desarrollo de tratamientos. Los investigadores están explorando terapias que refuercen los sistemas de defensa celular en las primeras etapas de la vida, con el objetivo de retrasar aún más la aparición de los síntomas.
- Terapias génicas: Estrategias para silenciar o corregir el gen mutado podrían detener la producción de huntingtina tóxica.
- Reforzar la autofagia: Medicamentos que estimulen los procesos de limpieza celular podrían reducir la acumulación de proteínas defectuosas.
- Prevención del estrés oxidativo: Antioxidantes dirigidos podrían proteger a las neuronas del daño mitocondrial.
Mirando hacia el futuro
Aunque la enfermedad de Huntington sigue siendo incurable, los avances en la comprensión de su progresión lenta han renovado la esperanza en la comunidad científica y en las familias afectadas. Cada descubrimiento acerca de los mecanismos celulares y los factores que retrasan su manifestación nos acerca más a desarrollar estrategias que puedan prolongar la vida y mejorar la calidad de las personas con esta condición.
Con investigación constante y colaboraciones globales, la pregunta ya no es si podremos detener la enfermedad de Huntington, sino cuándo.
Temas:
- Cerebro
- Enfermedades
Lo último en OkSalud
-

Ni gorras ni gafas de sol: la prenda de vestir que recomiendan los expertos a los mayores de 65 años para evitar los efectos del calor en verano
-
No compres gafas para el eclipse si no llevan este símbolo escrito: es un requisito para su homologación
-
¿Qué significa cuando alguien repite «gracias» según la psicología?
-
Unos segundos mirando el eclipse pueden bastar para perder visión de forma permanente
-
Adiós al alcohol: la OMS pide a la UE subir impuestos y limitar su publicidad para evitar 10.000 cánceres





Últimas noticias
-
La Premier vuelve a sonrojar a la Liga de Tebas y ya la quintuplica en gastos en fichajes
-
Entrada de inmigrantes a Ceuta desde Marruecos, en directo: fecha de la nueva avalancha, situación en la frontera y reacciones
-
La Policía Nacional detiene al proetarra que agredió a Cake Minuesa en Bilbao durante la final del Mundial
-
Blatter se moja sobre la oposición a Infantino: «Es el momento de que una mujer asuma el liderazgo de la FIFA»
-
Ni un mena más




