Un consorcio internacional identifica nuevos genes de resistencia a los antibióticos en la tuberculosis
Fact checked
Este artículo de OkSalud ha sido verificado para garantizar la mayor precisión y veracidad posible: se incluyen, en su mayoría, estudios médicos, enlaces a medios acreditados en la temática y se menciona a instituciones académicas de investigación. Todo el contenido de OkSalud está revisado pero, si consideras que es dudoso, inexacto u obsoleto, puedes contactarnos para poder realizar las posibles modificaciones pertinentes.
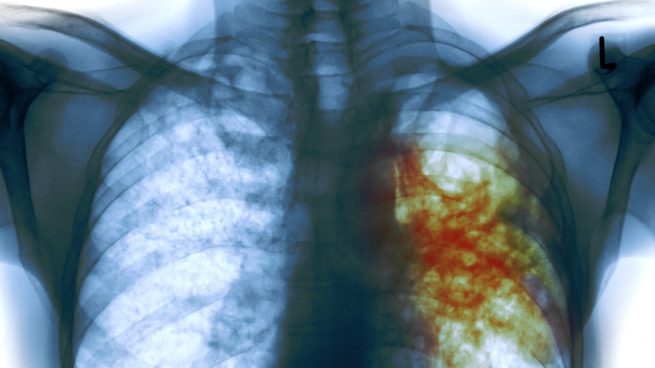
Un análisis masivo de más de 10.000 aislados diferentes de la bacteria Mycobacterium tuberculosis procedentes de 23 países ha revelado nuevos genes asociados a la resistencia a 13 antibióticos de primera y segunda línea, tanto nuevos como reutilizados.
El trabajo, llevado a cabo por el Comprehensive Resistance Prediction for Tuberculosis: an International Consortium (CRyPTIC), se describe en dos nuevos artículos publicados en la revista de acceso abierto ‘PLOS Biology’.
La tuberculosis (TB) es una enfermedad curable y prevenible ya que el 85% de los afectados pueden ser tratados con éxito con un régimen de medicamentos de seis meses. A pesar de ello, ha matado a más personas que otras enfermedades infecciosas en muchos años recientes, y la TB resistente a los medicamentos es una amenaza continua. Por ello, conocer mejor las variantes de ‘M. tuberculosis’ que confieren resistencia a los antibióticos es importante tanto para un mejor seguimiento de las cepas resistentes como para el desarrollo de nuevos fármacos.
En el primer artículo, los investigadores describen cómo han reunido un compendio de datos de libre acceso de 12.289 aislados de ‘M. tuberculosis’, procesados en laboratorios asociados de CRyPTIC de todo el mundo. Cada aislado se secuenció y luego se probó en una parrilla de alto rendimiento con concentraciones variables de 13 antimicrobianos. De las muestras incluidas en el compendio, 6.814 eran resistentes a al menos un fármaco, incluidas 4.685 muestras resistentes a varios fármacos o al tratamiento de primera línea, la rifampicina.
En el segundo artículo, el consorcio presentó los resultados de un estudio de asociación del genoma (GWAS) con los datos de 10.228 aislados de ‘M. tuberculosis’. Para los 13 fármacos, el grupo descubrió variantes no catalogadas asociadas a aumentos significativos de la concentración mínima inhibitoria, es decir, la concentración más baja de un antibiótico que detiene el crecimiento de ‘M. tuberculosis’.
El análisis de esta concentración, en lugar de un resultado binario de resistente o no resistente, permitió identificar variantes que sólo causan cambios sutiles en la respuesta al antibiótico que pueden superarse aumentando la dosis del fármaco. Los investigadores seleccionaron los 20 genes más significativos que confieren resistencia a cada fármaco y describieron con mayor profundidad el tamaño del efecto y las variaciones dentro de estos genes específicos.
«Nuestro estudio demuestra la capacidad de las asociaciones mundiales para mejorar sustancialmente nuestro conocimiento de las variantes genéticas asociadas a la resistencia a los antimicrobianos en ‘M. tuberculosis’», señalan los autores.
En conjunto, los trabajos no sólo descubren genes específicos que pueden ser objeto de seguimiento para comprender mejor el panorama de la resistencia de ‘M. tuberculosis’, sino también un marco para futuros estudios sobre el patógeno.
«El compendio no está diseñado para medir la prevalencia o estimar las tasas de error ‘en el mundo real’ de las herramientas de predicción de la resistencia; más bien sirve como recurso para acelerar el desarrollo del diagnóstico de la resistencia a los antimicrobianos mediante el enriquecimiento de los catálogos de mutaciones para la predicción de la resistencia de la secuenciación del genoma completo, la mejora de nuestra comprensión de los mecanismos genéticos de la resistencia y la identificación de importantes lagunas de diagnóstico y patrones de resistencia a los medicamentos -dicen los autores-. El compendio de datos es totalmente de código abierto y se espera que facilite e inspire futuras investigaciones durante años».
Lo último en OkSalud
-

Ni avena ni centeno: el cereal superalimento que ayuda a prevenir la metástasis del cáncer, según expertos en medicina
-
Qué significa que una persona sea amable con todo el mundo pero no tenga amigos íntimos, según la psicología
-
Suicidio: principal causa de muerte en los jóvenes de entre 15 y 29 años
-
Ésta es la dieta favorita de los dermatólogos para cuidar la piel
-
Meritxell Martí: «El farmacéutico es el profesional indicado para recomendar un suplemento»




Últimas noticias
-
Caída de Márquez en Austin: se va al suelo en la primera vuelta de la sprint
-
El cargo del Gobierno que hizo de guía de Sánchez en el Pirineo promociona los negocios de Begoña
-
Bezzecchi y Marini, sancionados con dos posiciones de parrilla por su acción antideportiva con Márquez
-
El Málaga se atasca y el Leganés saca petróleo de La Rosaleda
-
Comprobar ONCE hoy, sábado, 28 de marzo de 2026: Sueldazo y Super 11




